En una entrada anterior informábamos que cerca de la mitad de los casos de discapacidad intelectual permanecen sin diagnostico etiológico y que se estima que un 40% tienen origen genético. Por su mayor frecuencia destaca la trisomía 21 y el síndrome de Xfrágil. Sin embargo, la casuística es muy amplia y en la plataforma de genes y fenotipos relacionados con la discapacidad intelectual Orphanet y ERN ITHACApodemos encontrar 979 enfermedades genéticas raras que cursan con discapacidad intelectual.
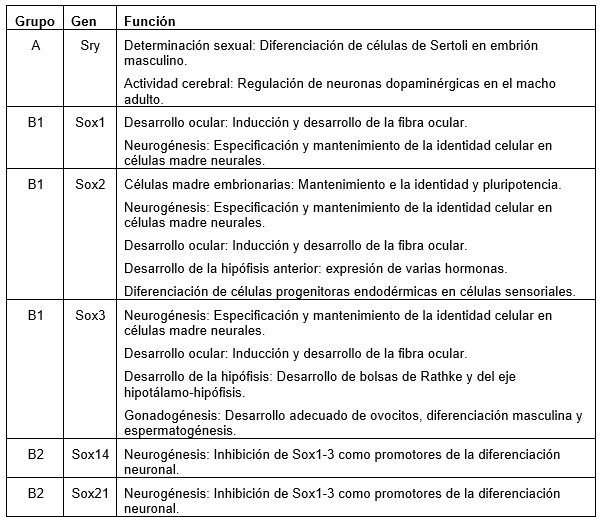
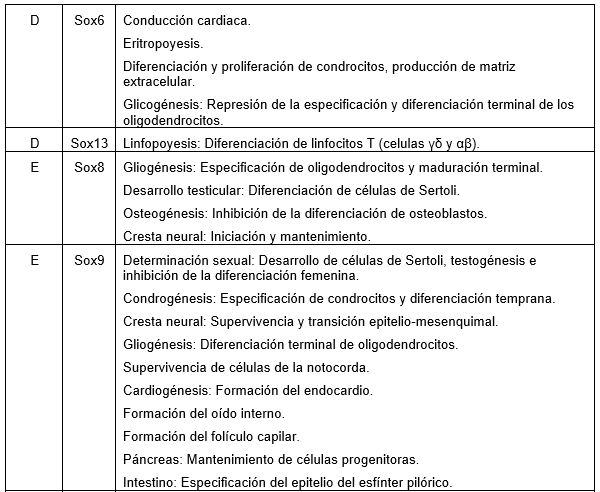
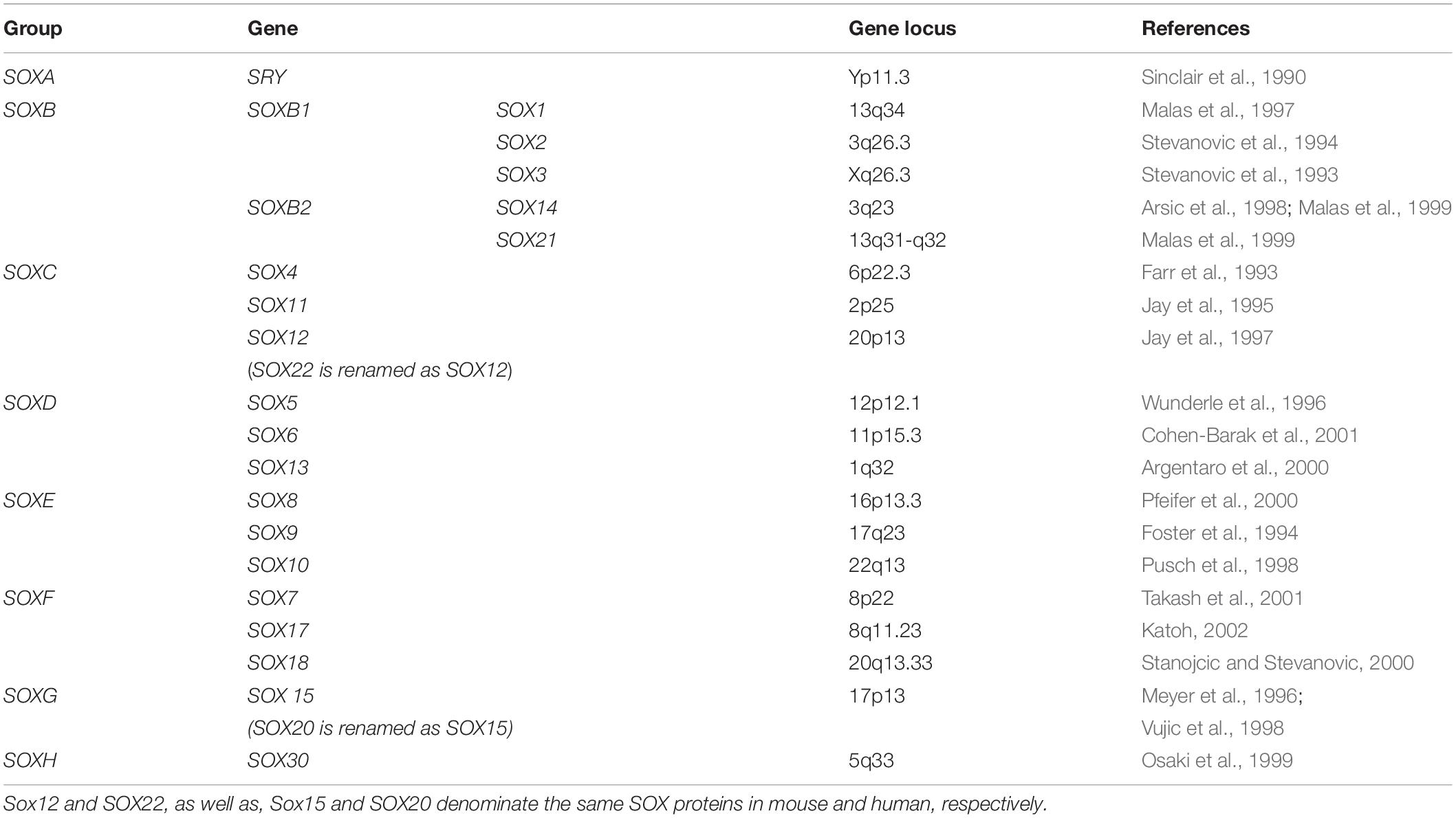
Las proteínas SOX pertenecen a la familia de los factores de transcripción (Tfs) poseyendo un dominio de unión al ADN de la clase HMG. Se codifican desde los genes Sox y tienen funciones en el desarrollo de órganos y destino celular. En Leon, A. (2019) podemos encontrar el grupo y las funciones de cada uno de los genes Sox (1).
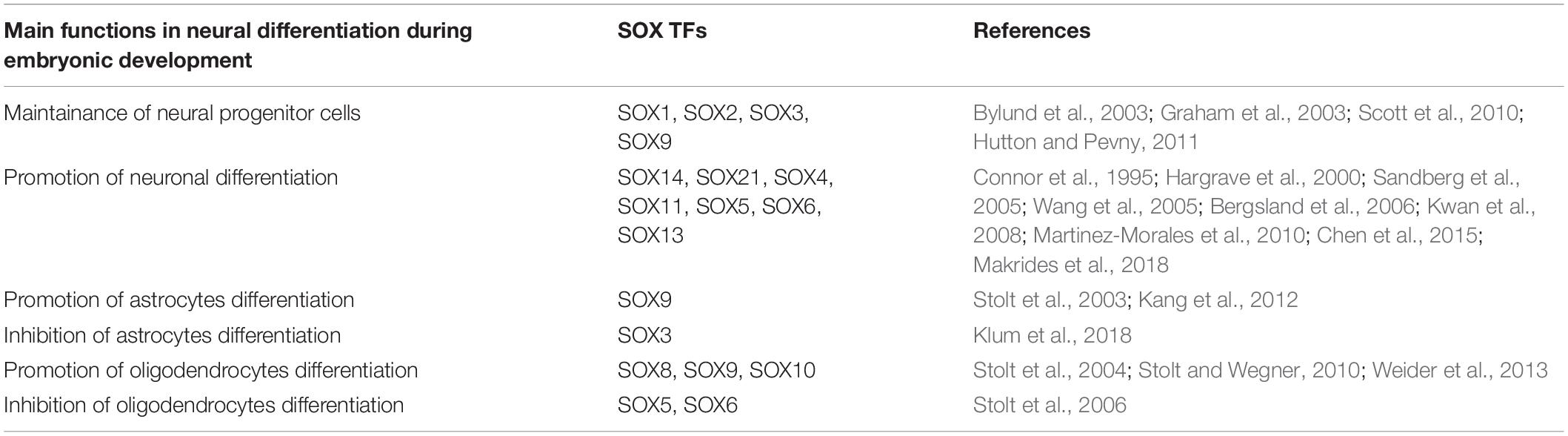
(Clic para ampliar las imágenes) Por su parte Stevanovic et al. (2021) proporcionan una síntesis de sus implicaciones para el desarrollo del sistema nervioso y la neurogénesis adulta y la localización de cada gen (2).
(Clic para ampliar la imagen)
Dada su implicación en la configuración del SN no es de extrañar que alteraciones genéticas en ellos puedan conllevar discapacidad intelectual. El ya citado trabajo de Stevanovic et al. (2021) proporciona un resumen de las variantes asociadas a trastornos del neurodesarrollo (Tabla suplementaria S1). En Underwood et al., podemos encontrar un revisión de las variantes y fenotipos asociados (3).
2.- Stevanovic M, Drakulic D, Lazic A, Stanisavljevic Ninkovic D, Schwirtlich M and Mojsin M (2021) SOX Transcription Factors as Important Regulators of Neuronal and Glial Differentiation During Nervous System Development and Adult Neurogenesis. Front. Mol. Neurosci. 14:654031. doi: 10.3389/fnmol.2021.654031. https://www.frontiersin.org/articles/10.3389/fnmol.2021.654031/full
3.- Underwood, A.; Rasicci,D.T.; Hinds, D.; Mitchell, J.T.; Zieba,J.K.; Mills, J.; Arnold, N.E.; Cook,T.W.; Moustaqil, M.; Gambin, Y.; et al.Evolutionary Landscape of SOXGenes to Inform Genotype-to-Phenotype Relationships.Genes2023,14, 222. https://doi.org/10.3390/genes14010222
Por una entrada de Gen-Ética hemos sabido de la Foundation29. Su apuesta por los estandares abiertos, la recopilación y compartición de datos y la inteligencia artificial son aspectos a destacar. En este ámbito proporcionan tres herramientas:
1.- Dx29 permite la introducción de síntomas y la obtención de posibles diagnósticos asociados. De forma inversa permite a partir de un diagnóstico obtener los síntomas y signos asociados. Si bien la página de Orphanet provee de información parecida destaca la amigabilidad de la interface. Claramente mejora nuestra aproximación al tema de la clínica.
2.- HealthData29 proporciona una plataforma para que las instituciones «pongan sus datos abiertos a disposición de la comunidad con fines de investigación. Se realiza a través del acceso controlado, garantizando la seguridad de los datos y el respeto de los pacientes».
3.- Raito está dirigida a pacientes permitiendo que creen un deposito de su información y permite compartirlos con los médicos, asociaciones de pacientes o investigadores.
Igualmente disponen de Lab29 en que trabajan con la inteligencia artificial y cuyos proyectos actuales son:
1.- DxGPT “es un experimento pionero que utiliza grandes modelos de lenguaje para crear sistemas de apoyo al diagnóstico médico. Esta aplicación explora cómo la IA puede asistir en la identificación precisa y rápida de condiciones médicas, mejorando el proceso de diagnóstico y el cuidado del paciente”.
2.- Nav29 “representa nuestro esfuerzo por utilizar agentes inteligentes para ayudar a los pacientes en tareas específicas. Entendemos que el cuidado de la salud no se limita a los hospitales, sino que es una parte integral de la vida diaria del paciente. Nav29 está diseñado para asistir en actividades como la búsqueda de ensayos clínicos, información sobre medicamentos y datos científicos actualizados, actuando como un navegador o acompañante a lo largo de la enfermedad”.
3.- Summary29 “es un conjunto de herramientas destinadas a resumir información médica compleja. Este proyecto busca aliviar la carga de los profesionales médicos y pacientes en la comprensión y gestión de datos médicos, simplificando la información de informes y conversaciones médicas. Nuestra meta es hacer accesible la información médica, reduciendo el tiempo y los recursos necesarios para su procesamiento”.
El 04/10/2022 el Centro de Investigación Biomédica en Red (CIBER) informaba de la nueva plataforma OrphaID que permite “acceso sistemático a la información específica de Orphanet sobre los genes relacionados con la discapacidad intelectual y los fenotipos asociados…La plataforma, disponible en inglés, proporciona información que se puede filtrar por nombre de gen, trastorno, código ORPHA, etc., con una serie de formatos propuestos para descargar los resultados de la búsqueda».
Tras probarla hemos querido reseñarla dado que supone una forma ágil de acceder a la información de una determinada entidad o de entidades relacionadas con un término HPO.
La discapacidad intelectual pueden originarse por una gran número de causas que, según el momento, se clasifican en:
1.- Prenatales: en la propia concepción por causa hereditaria o alteración en la meiosis de las células germinales paternas o maternas (genética) o por afectación del embrión por causa externa (teratógenos).
2.- Perinateles: acontecimientos que se dan durante el trabajo del parto.
3.- Neonatales: son factores de riesgo que se dan durante las cuatro primeras semanas de vida (daño cerebral, encefalopatía, convulsiones neonatales, sepsis, hemorragia y apnea).
4.- Postnatales: cualquier afectación del sistema nervioso central que se da antes de los 18 años.
Independientemente de la causa la constante la constituye una alteración cerebral que afecta a las funciones intelectuales que pueden manifestarse en distintos grados de afectación de cada uno de sus componentes o funciones ejecutivas.
Tal como índica su título, està entrada pretende ser, como define la Real Academia, “un libro de poco volumen y de fácil manejo para consulta inmediata de nociones o informaciones fundamentales” sobre enfermedades que cursan con discapacidad intelectual.
A partir de la infomación que permite descargar Orphanet hemos creado una tabla con las diferentes enfermedades que cursan con discapacidad intelectual. Incluye enlaces a cada página Orphanet en que se amplia información sobre ellas.
En una entrada anterior realizamos una relación de los principales sistemas diagnósticos para los Trastornos del Espectro Alcohólico Fetal (TEAF/FASD) Si bien tal relación no es exhaustiva sí es suficiente para evidenciar que no hay unos criterios diagnósticos universales. En esta entrada vamos a realizar una comparativa de las distintas guías centrándonos en las versiones más recientes.
1.- En primer lugar encontramos diferencias terminológicas:
Sistema diagnóstico
Categorías diagnósticas
IOM
SAF/FAS – SAFP/FASP – ARND- ARBD
4-Digit Diagnostic Code
22 categorías diagnósticas (de la A a la V)
Canadian diagnostic guideline
FASD con rasgos faciales – FASD sin rasgos faciales – Riesgo de FASD
CDC
SAF/FAS
Emory-20
SAF/FAS – SAFP/FASP – ARND- Exposición prenatal sin diagnóstico.
Australian Guide
FASD con 3 rasgos faciales – FASD con menos de 3 rasgos faciales.
German guideline
SAF/FAS
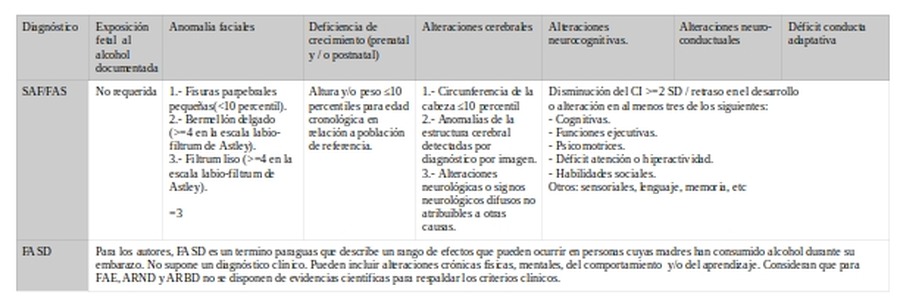
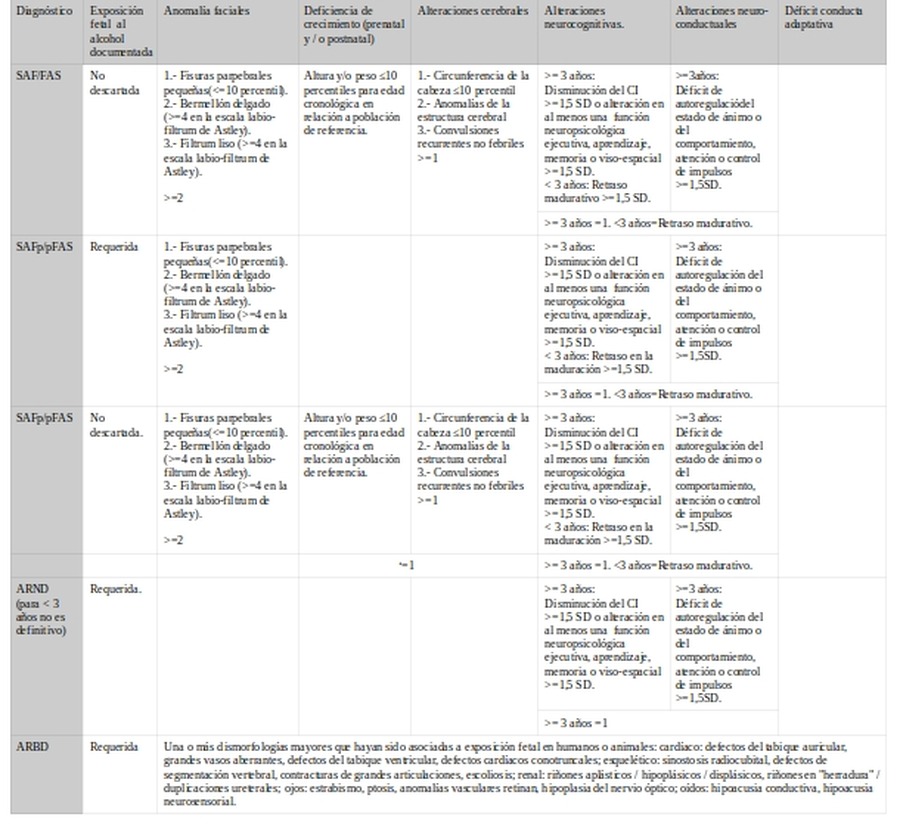
2.- Se encuentran igualmente diferencias en los criterios para los signos o síntomas a valorar.
a.- Exposición prenatal al alcohol (EPA/PAE)
EPA/PAE
SAF/FAS
SAFp/pFAS
ARND
ARBD
IOM-2016
1.- Confirmada.
2.- No conocida.
3.- No.
Fuentes: Cuestionario AUDIT, T-ACE, información de la madre y otros (terceros incluidos).
No requerida
Requerida o no descartada (según subtipo)
Requerida
Requerida
4-Digit
1. Se ha confirmado ningún consumo durante embarazo.
2. No conocido: no se tiene información o no es fiable.
3. Riesgo: consumo confirmado con un valor y frecuencia inferior al nivel 4 o no se conoce su nivel.
4.- Alto riesgo: se ha confirmado el consumo con altas cantidades al menos una vez a la semana al principio del embarazo.
Fuentes: Información de la madre y otros (terceros incluidos).
Nivel 3-4 o nivel 2 (según subtipo)
Nivel 3-4
Nivel 3-4
Nivel 3-4
Canadian
1.- Sí.
2.- No.
3.- No conocida.
Fuentes: Cuestionario CRAFFT, TWEAK, información de la madre y otros (terceros incluidos).
Requerida o no descartada
Requerida
Requerida
Requerida
CDC
1.- Confirmada.
2.- No conocida.
Fuentes: Cuestionario AUDIT, T-ACE, TWEAK, MAST, S-MAST, NET, RAPS4, RAPS4-QF, información de la madre y otros (terceros incluidos).
No requerida
Emory-20
1.- Sí. (no se admite información de terceros)
2.- No.
3.- No conocida.
No requerida
Requerida
Requerida
Australian Guide
1.- No.
2.- No conocido.
3.- Exposición.
4.- Exposición de alto riesgo.
Fuentes: Cuestionario AUDIT-C, información de la madre y otros (terceros incluidos).
No requerida
Requerida
German guideline
No se requiere si el fenotipo facial está presente,
No requerida
En la comparativa puede observarse que hay acuerdo en relación a que salvo para SAF/FAS se va a requerir que se haya confirmado el consumo de alcohol durante la gestación (EPA/PAE). Para el fenotipo completo (SAF/FAS) se asume que se ha dado el consumo.
b.- Anomalías faciales
Anomalías faciales
SAF/FAS
SAFp/pFASp
ARND
ARBD
IOM-2016
1.- Fisuras parpebrales pequeñas(<=10 percentil).
2.- Bermellón delgado (>=4 en la escala labio-filtrum de Astley).
3.- Filtrum liso (>=4 en la escala labio-filtrum de Astley).>=2
1.- Fisuras parpebrales pequeñas(<=10 percentil).
2.- Bermellón delgado (>=4 en la escala labio-filtrum de Astley).
3.- Filtrum liso (>=4 en la escala labio-filtrum de Astley).>=2
4-Digit
1.- Fisuras parpebrales pequeñas (<2DS <3 percentil).
2.- Bermellón delgado (>=4 en la escala labio-filtrum de Astley).
3.- Filtrum liso (>=4 en la escala labio-filtrum de Astley).=3
1.- Fisuras parpebrales pequeñas (<2DS <3 percentil).
2.- Bermellón delgado (>=4 en la escala labio-filtrum de Astley).
3.- Filtrum liso (>=4 en la escala labio-filtrum de Astley).=3
Canadian-2015
FADS con rasgos faciales
FADS sin rasgos faciales
1.- Fisuras parpebrales pequeñas(<=-2SD <3 percentil).
2.- Bermellón delgado (>=4 en la escala labio-filtrum de Astley).
3.- Filtrum liso (>=4 en la escala labio-filtrum de Astley).=3
1.- Fisuras parpebrales pequeñas(<=-2SD <3 percentil).
2.- Bermellón delgado (>=4 en la escala labio-filtrum de Astley).
3.- Filtrum liso (>=4 en la escala labio-filtrum de Astley).
o
1.- Fisuras parpebrales pequeñas(>-2SD y <=-1SD).
2.- Bermellón delgado (>=4 en la escala labio-filtrum de Astley).
3.- Filtrum liso (>=4 en la escala labio-filtrum de Astley).
o
1.- Fisuras parpebrales pequeñas(<=-2SD <3 percentil).
2.- Bermellón delgado (=3 en la escala labio-filtrum de Astley).
3.- Filtrum liso (>=4 en la escala labio-filtrum de Astley).
o
1.- Fisuras parpebrales pequeñas(<=-2SD <3 percentil).
2.- Bermellón delgado (>=4 en la escala labio-filtrum de Astley).
3.- Filtrum liso (=3 en la escala labio-filtrum de Astley).
CDC
1.- Fisuras parpebrales pequeñas(<10 percentil).
2.- Bermellón delgado (>=4 en la escala labio-filtrum de Astley).
3.- Filtrum liso (>=4 en la escala labio-filtrum de Astley).=3
Emory-20
Incluye un ckeck-list en que se valoran otros aspectos junto con fisuras parpebrales, bermellón y filtrum.
>10 efectos leves. >20 efectos graves.
Incluye un ckeck-list en que se valoran otros aspectos junto con fisuras parpebrales, bermellón y filtrum.
>10 efectos leves. >20 efectos graves. No son necesarios los 3 rasgos faciales.
Australian Guide
1.- Fisuras parpebrales pequeñas(<=-2SD <3 percentil).
2.- Bermellón delgado (>=4 en la escala labio-filtrum de Astley).
3.- Filtrum liso (>=4 en la escala labio-filtrum de Astley).=3
1.- Fisuras parpebrales pequeñas(<=-2SD <3 percentil).
2.- Bermellón delgado (>=4 en la escala labio-filtrum de Astley).
3.- Filtrum liso (>=4 en la escala labio-filtrum de Astley).<3
German guideline
1.- Fisuras parpebrales pequeñas(<=3er percentil).
2.- Bermellón delgado (>=4 en la escala labio-filtrum de Astley).
3.- Filtrum liso (>=4 en la escala labio-filtrum de Astley).=3
Para FAS/FAE se observa que hay acuerdo en que tanto bermellón como filtrum requieren de puntuación mayor o igual a 4 en escala de Astley. Sin embargo el punto de corte varía para fisuras y número de signos: Observamos que el criterio IOM se encuentra entre los más laxos.
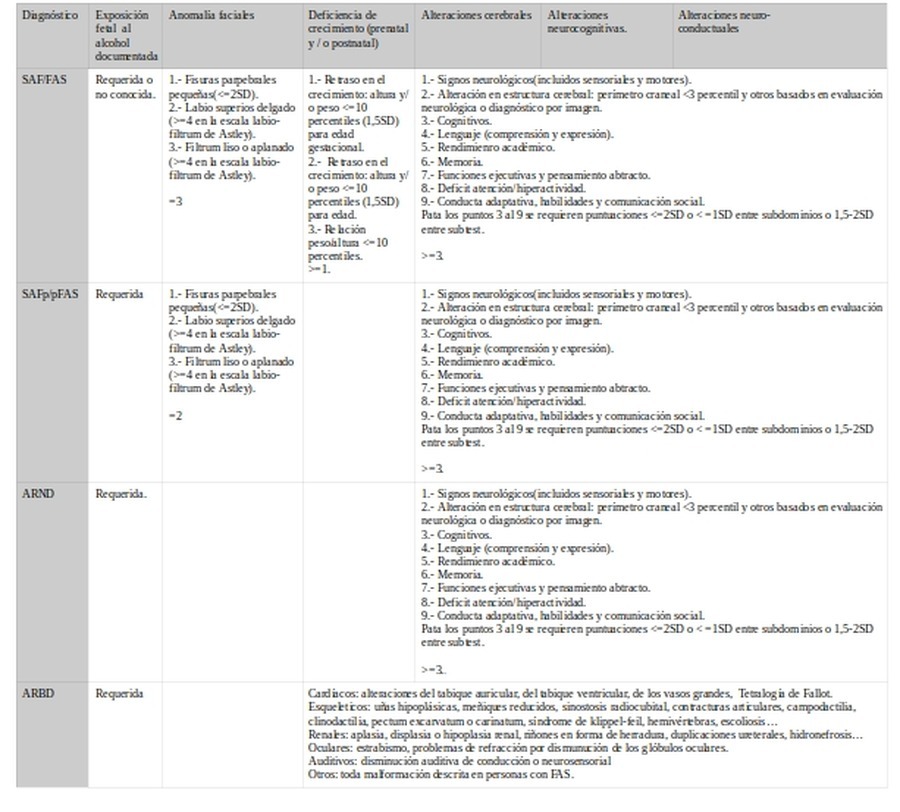
c.- Alteraciones del crecimiento.
Crecimiento
SAF/FAS
SAFp/pFASp
ARND
ARBD
IOM-2016
Altura y/o peso ≤10 percentiles para edad cronológica en relación a población de referencia.
Altura y/o peso ≤10 percentiles para edad cronológica en relación a población de referencia o no requerido
4-Digit
1.- Altura y/o peso <=3centiles
o
2.- Altura y peso >3 y <=10 centiles.
1.- Altura y/o peso <=3centiles
o
2.- Altura y peso >3 y <=10 centiles.
o
3.- Altura o peso >3 y <=10 centiles
o
no requerido.
Canadian-2015
FAS con rasgos faciales/sin rasgos faciales
No requerido
CDC
Altura y/o peso ≤10 percentiles para edad cronológica en relación a población de referencia.
Emory-20
Altura y/o peso ≤10 percentiles para edad cronológica en relación a población de referencia.
Altura y/o peso ≤10 percentiles para edad cronológica en relación a población de referencia o no requerido.
Australian Guide
No requerido
No requerido
German guideline
1.- Peso, al nacer o actual, ≤10 percentiles.
2.- Altura, al nacer o actual, <=10 percentiles.
3.- IMC<=10 percentiles.>=1.
El acuerdo está en <=10 percentiles. La excepción está en Canadian-2015 que no requiere tal variable.
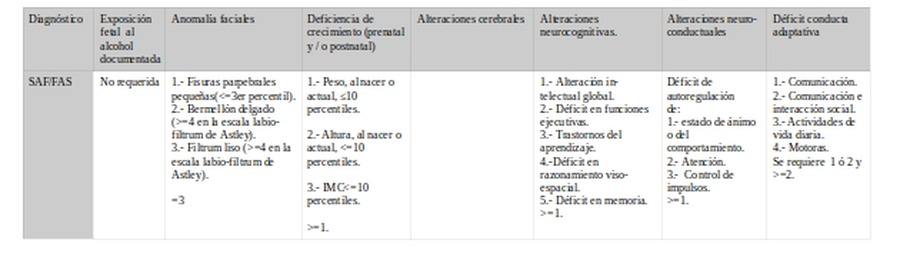
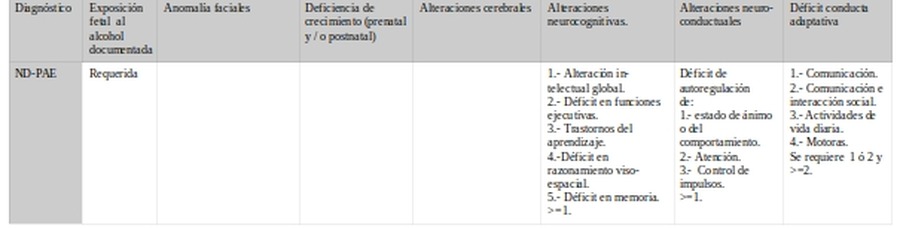
d.- Daño cerebral
Daño cerebral
Alteraciones cerebrales
Alteraciones neurocognitivas.
Alteraciones neuro-
conductuales
Déficit conducta adaptativa
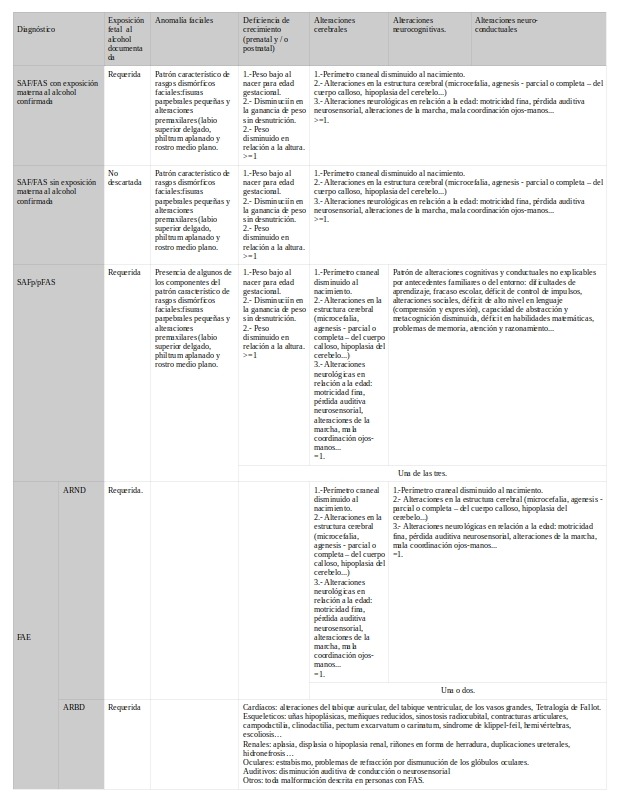
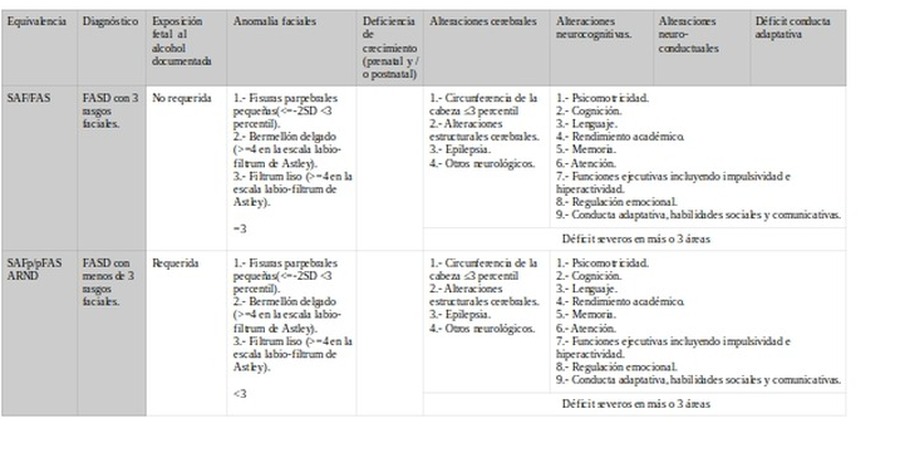
IOM-2016SAF/FAS SAFp/pFAS ARND >3años.
1.- Circunferencia de la cabeza ≤10 percentil
2.- Anomalías de la estructura cerebral
3.- Convulsiones recurrentes no febriles
>=1
>= 3 años:
Disminución del CI >=1,5 SD o alteración en al menos una función neuropsicológica ejecutiva, aprendizaje, memoria o viso-espacial >=1,5 SD.
< 3 años: Retraso madurativo >=1,5 SD.
>=3años:
Déficit de autoregulació del estado de ánimo o del comportamiento, atención o control de impulsos >=1,5SD.
>= 3 años =1. <3 años=Retraso madurativo
4-Digit
1.- Alteraciones significativas en tres o más funciones cerebrales tales como cognición, memoria, funciones ejecutivas, motricidad, lenguaje, atención actividad o signos neurológicos difusos.
2.- Microcefalia (COF<=-2DS 2,5 percentiles) y/o alteraciones significativas en en la estructura cerebral de probable origen prenatal y/o alteraciones neurológicas severas de probable origen prenatal.
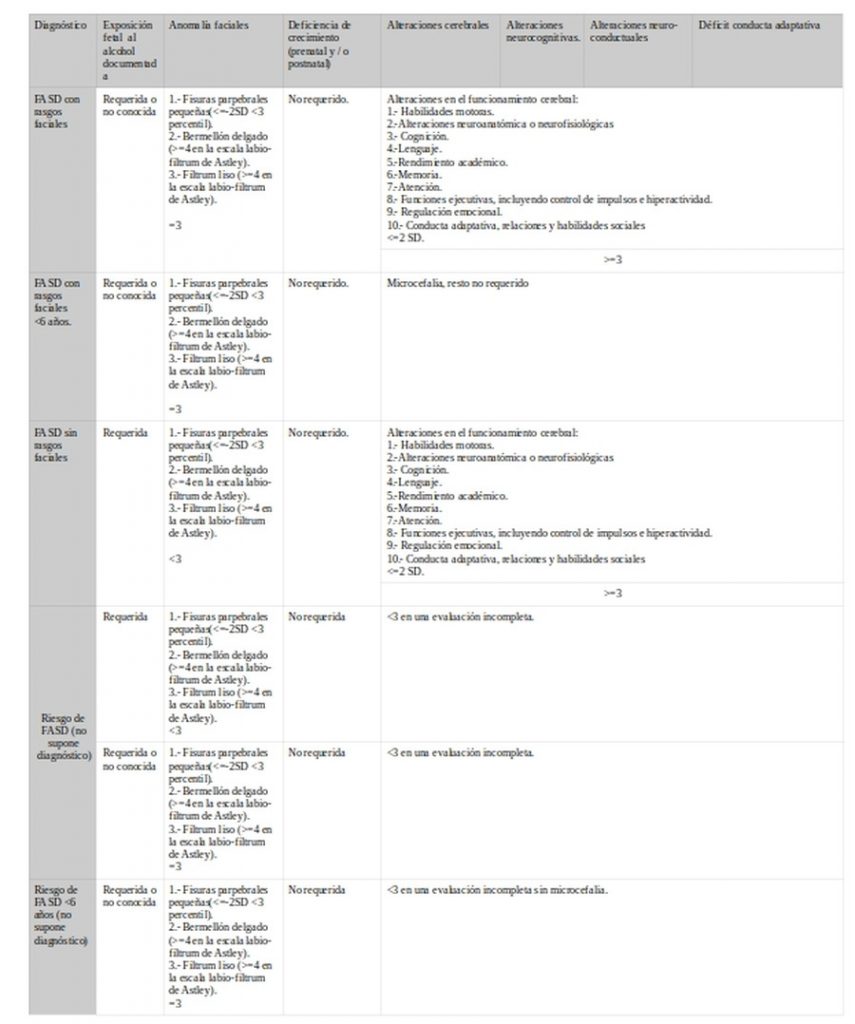
Canadian-2015
FAS con rasgos faciales/sin rasgos faciales.
Alteraciones en el funcionamiento cerebral:
1.- Habilidades motoras.
2.-Alteraciones neuroanatómica o neurofisiológicas
3.- Cognición.
4.-Lenguaje.
5.-Rendimiento académico.
6.-Memoria.
7.-Atención.
8.- Funciones ejecutivas, incluyendo control de impulsos e hiperactividad.
9.- Regulación emocional.
10.- Conducta adaptativa, relaciones y habilidades sociales
<=2 SD.
>=3
Para menores de 6 años: microcefalia, resto no requerido.
CDC
1.- Circunferencia de la cabeza ≤10 percentil
2.- Anomalías de la estructura cerebral detectadas por diagnóstico por imagen.
3.- Alteraciones neurológicas o signos neurológicos difusos no atribuibles a otras causas.
Disminución del CI >=2 SD / retraso en el desarrollo o alteración en al menos tres de los siguientes:
– Cognitivas.
– Funciones ejecutivas.
– Psicomotrices.
– Déficit atención o hiperactividad.
– Habilidades sociales.
Otros: sensoriales, lenguaje, memoria, etc
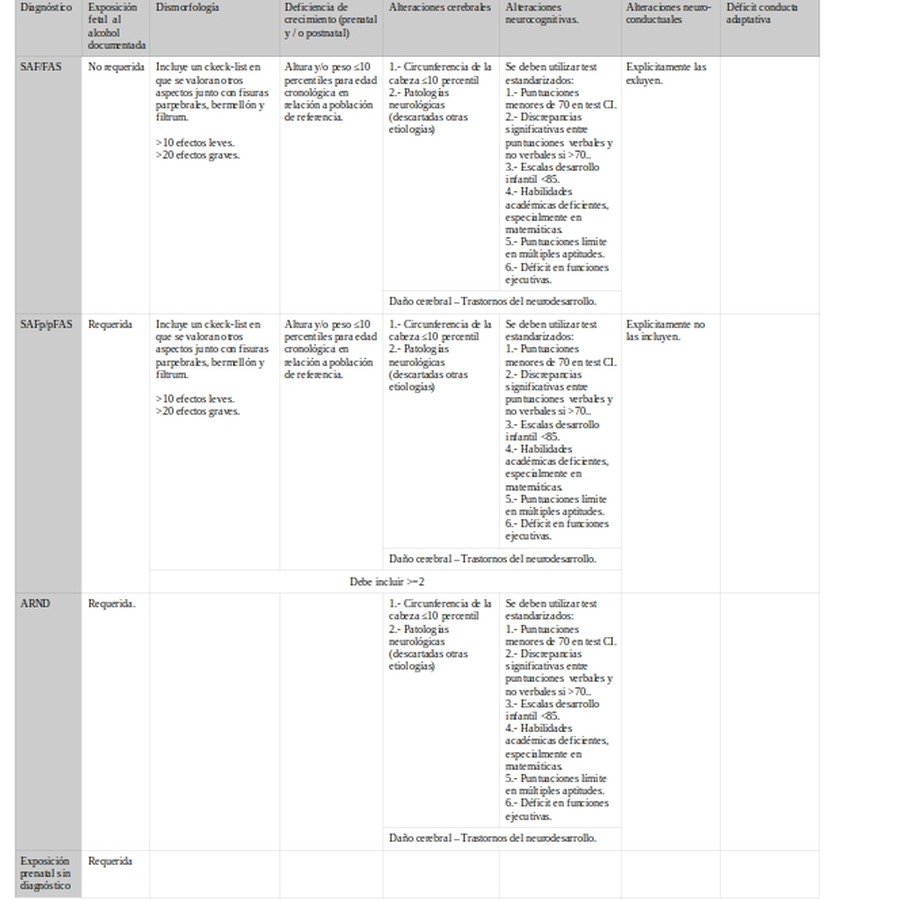
Emory-20
SAF/FAS
SAFp/pFAS
ARND
1.- Circunferencia de la cabeza ≤10 percentil
2.- Patologías neurológicas (descartadas otras etiologías)
Se deben utilizar test estandarizados:
1.- Puntuaciones menores de 70 en test CI.
2.- Discrepancias significativas entre puntuaciones verbales y no verbales si >70..
3.- Escalas desarrollo infantil <85.
4.- Habilidades académicas deficientes, especialmente en matemáticas.
5.- Puntuaciones límite en múltiples aptitudes.
6.- Déficit en funciones ejecutivas.
Explícitamente las excluyen.
Australian Guide
FADS con/sin rasgos faciales
1.- Circunferencia de la cabeza ≤3 percentil
2.- Alteraciones estructurales cerebrales.
3.- Epilepsia.
4.- Otros neurológicos.
1.- Psicomotricidad.
2.- Cognición.
3.- Lenguaje.
4.- Rendimiento académico.
5.- Memoria.
6.- Atención.
7.- Funciones ejecutivas incluyendo impulsividad e hiperactividad.
8.- Regulación emocional.
9.- Conducta adaptativa, habilidades sociales y comunicativas.
Déficit severos en más o 3 áreas
German guideline
1.- Alteración intelectual global.
2.- Déficit en funciones ejecutivas.
3.- Trastornos del aprendizaje.
4.-Déficit en razonamiento viso-espacial.
5.- Déficit en memoria.
>=1.
Déficit de autoregulación
de:
1.- estado de ánimo o del comportamiento.
2.- Atención.
3.- Control de impulsos.
>=1.
1.- Comunicación.
2.- Comunicación e interacción social.
3.- Actividades de vida diaria.
4.- Motoras.
Se requiere 1 ó 2 y >=2.
El punto de acuerdo se encuentra en la existencia de alteraciones cerebrales. Variará en los criterios de las manifestaciones cognitivas y conductuales y en el punto de corte.
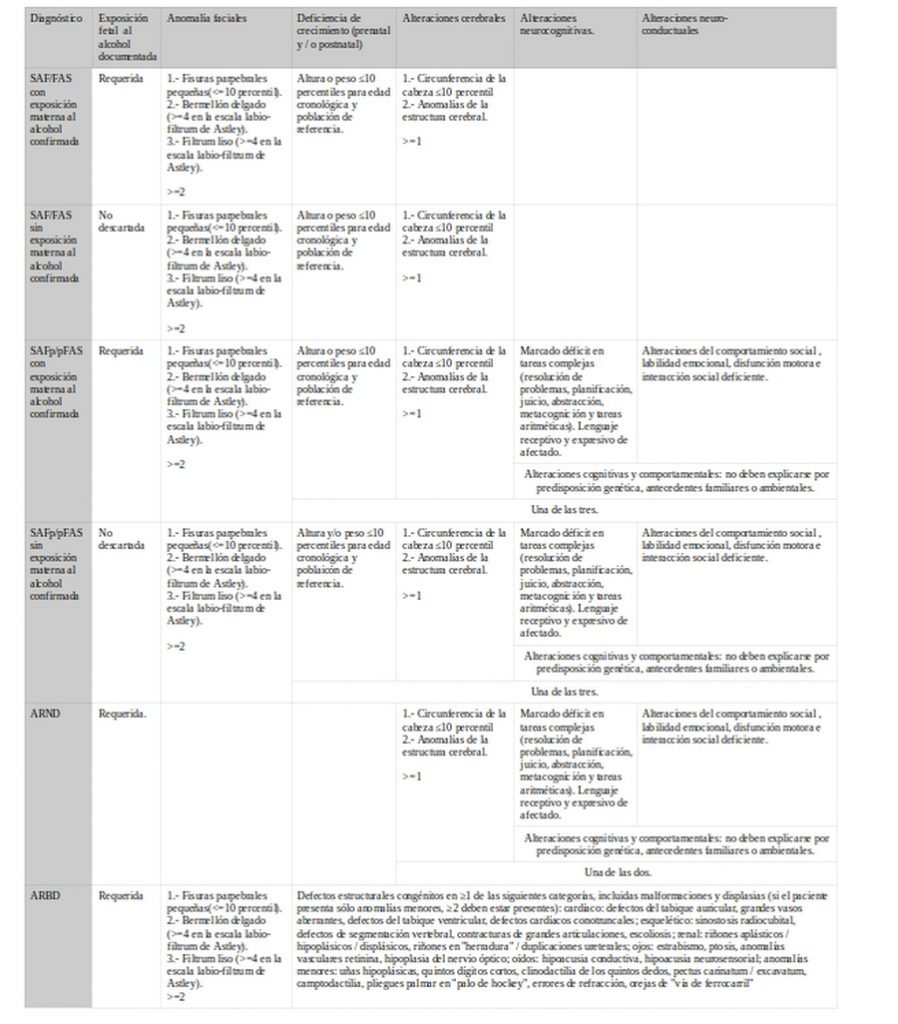
En conjunto podemos observar que una de las mayores dificultades de este conjunto de condiciones es que no presentan signos patognósticos y su único signo característico lo constituyen los rasgos faciales para SAF/FAS. Otra complicación se da por el hecho de que los signos son antropo o psicométricos y se han de valorar de acuerdo a la variabilidad del grupo de referencia o familiares. En los que se refiere al aspecto antropométrico nos parece muy ilustrativo el debate que se establece entre Astley1 y Coles et al2.
Coles et al1, en el trabajo que dio lugar al debate señalado, nos muestran que estas diferencias no son neutras. Con datos de 1.581 casos entre 0 y 21 años se aplicaron distintos criterios diagnósticos obteniéndose los siguientes resultados. En este trabajo también se puede encontrar una tabla comparativa de los distintos criterios con mayor profundidad que las que hemos presentado y que resulta de interés si se quiere profundizar en el tema.
Igualmente proporcionan el índice de Kappa para valorar el acuerdo entre los distintos sistemas. En la tabla siguiente mostramos los índices para SAF/FAS: en color rojo índices, según escala de Landis y Koch, insignificantes y azul para moderados.
Kappa
Emory-20
Emory-10
4-Digits 2.004
Canada 2.005
IOM 2.005
CDC
Emory-20
0,517
0,022
0,16
0,535
0,329
Emory-10*
0,517
0,007
0,063
0,369
0,17
4-Digits 2.004
0,022
0,007
0,117
0,022
0,097
Canada 2.005
0,16
0,063
0,117
0,172
0,506
IOM 2.005
0,535
0,369
0,036
0,172
0,407
CDC
0,329
0,17
0,097
0,506
0,407
Emory-10 utiliza como punto de corte 10 puntos en el check-list de alteraciones físicas.
Los promedios de acuerdo observados son 0,234 para SAF/FAS. Este acuerdo llega a 0,570 si se compara con recibir algún diagnóstico (SAF/FAS-SAFp/pFAS/ARND) o no recibirlo (se excluye CDC). La comparación en acuerdo para los distintos componentes 0,901 (crecimiento), 0,246 (facial) y 0,287 (neurocomportamental) nos muestra el bajo acuerdo incluso para los signos físicos característicos.
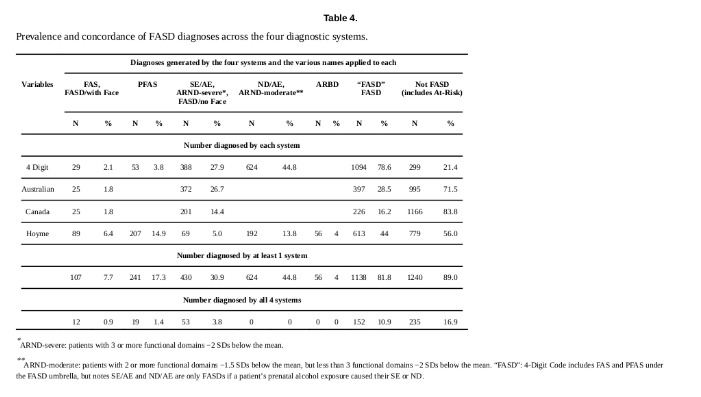
Por su parte Astley et al4 compararon 4-Digit 2.004, Canadian 2.015, IOM 2.016 y Australian 2.016 en 1.392 casos. Al igual que en el trabajo de Coles et al1 también se puede encontrar una tabla comparativa de los distintos criterios con mayor profundidad que las que hemos presentado y que resulta de interés si se quiere profundizar en el tema.
Observamos que el 81,8% recibieron diagnóstico de TEAF/FASD por algún sistema, pero sólo el 10,9% para todos ellos. Para SAF/FAS tenemos el 7,7% para algún sistema y 0,9% para todos.
Para Brown et al5 la ausencia de criterios unificados tiene implicaciones en la investigación, la práctica clínica y las políticas sanitarias. Consideran que hay evidencias limitadas o contradictorias que apoyen unos u otros criterios y que serán las medidas objetivas del momento y nivel de exposición las que van a permitir superar esta situación.
1.- Astley, S. (2017). Letter to the Editor Regarding Coles, Gailey, Mulle, Kable, Lynch, and Jones (2016): A Comparison Among 5 Methods for the Clinical Diagnosis of Fetal Alcohol Spectrum Disorders. Alcoholism: Clinical and Experimental Research, 41(1), 216-218.
2.- Coles, C. D., Gailey, A., Mulle, J., Kable, J. A., Lynch, M. E., & Jones, K. L. (2017). Response to Astley’s Letter to the Editor. Alcoholism, clinical and experimental research, 41(1), 219.
3.- Coles, C. D., Gailey, A. R., Mulle, J. G., Kable, J. A., Lynch, M. E., & Jones, K. L. (2016). A comparison among 5 methods for the clinical diagnosis of fetal alcohol spectrum disorders. Alcoholism: Clinical and Experimental Research, 40(5), 1000-1009.
4.- Astley, S. J. A., Bledsoe, J. M., Brooks, A., Davies, J. K., Jirikowic, T., Olson, E., & Thorne, J. C. (2019). Comparison of the 4-Digit Code, Canadian 2015, Australian 2016 and Hoyme 2016 fetal alcohol spectrum disorder diagnostic guidelines. Advances in pediatric research, 6(2).
5.- Brown, J. M., Bland, R., Jonsson, E., & Greenshaw, A. J. (2019). The standardization of diagnostic criteria for fetal alcohol Spectrum disorder (FASD): implications for research, clinical practice and population health. The Canadian Journal of Psychiatry, 64(3), 169-176.
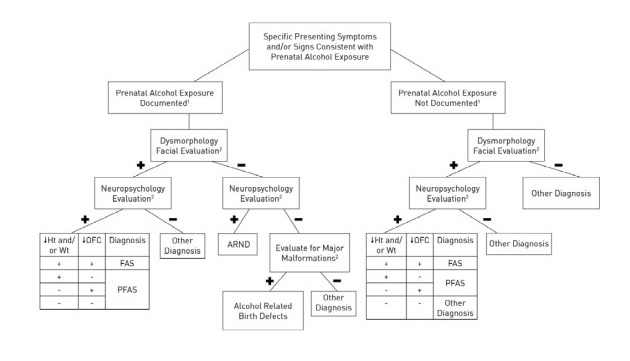
Tal y como informábamos en la primera entrada de esta categoría, bajo la denominación de Trastornos del Espectro Alcohólico Fetal (TEAF/FASD) se agrupan un conjunto de síntomas y signos que se presentan como consecuencia de la exposición prenatal al alcohol (EPA/PAE). En ella incluíamos la clasificación más utilizada. Sin embargo no es la única ya que se han establecido distintos criterios y categorizaciones sin haberse llegado a unificar yque vamos a resumir en esta nueva entrada:
1.- Standardized definition for FAS (Rosett-1980). 2.- Gestalt method (Sokol-1.989). 3.- The Institute of Medicine (IOM) -Stratton 1.996. 4.- Diagnostic Guide for Fetal Alcohol Syndrome and Related Conditions: The 4-Digit Diagnostic Code 1.999. 5.- Emory-20 2.000. 6.-Diagnostic Guide for Fetal Alcohol Syndrome and Related Conditions: The 4-Digit Diagnostic Code 2004. 7- Centers for Disease Control and Prevention (CDC)-2.004. 8.- Canadian diagnostic guideline 2.005. 9.- The Institute of Medicine (IOM) –Hoyme 2005. 10.- German guideline version 2013. 11.- Canadian diagnostic guideline 2015. 12.- Australian Guide to the diagnosis of Fetal Alcohol Spectrum Disorder (FASD)-2016. 13.- The Institute of Medicine (IOM) -Hoyme 2016. 14.- Mattson et al. -2019: The Institute of Medicine (IOM) -Hoyme 2016 + Trastornos Neurocomportamentales Relacionados con la Exposición Prenatal al Alcohol (ND-PAE)-DSM5.
1.- Standardized definition for FAS (Rosett-1980)1. Fue desarrollada por el Fetal Alcohol Study Group de la Research Society on Alcoholism (RSA). La RSA, a su vez, se enmarca en el National Institute on Alcohol Abuse and Alcoholism (NIAAA) y este es parte del National Institutes of Health (NIH). Por su afiliación entendemos que puede considerarse como precursos de las del del Institute of Medicine (IOM) o, como mínimo, que pasará a estar incluida en la Institute of Medicine (IOM) -Hoyme 2016. En consulta de 10/09/2020, Google Académico refiere 271 citas.
2.- Gestalt method (Sokol-1.989)2 Fue realizado por el Departamento de Obstetricia / Ginecología, Wayne State University / Hutzel Hospital Detroit, Michigan y la División de Estudios de Defectos Congénitos, Universidad de Washington, Seattle, Washington. Los autores proponen que estos criterios deben substituir los de Rosett-1980. Por sus orígenes entendemos que es el precursos de Diagnostic Guide for Fetal Alcohol Syndrome and Related Conditions: The 4-Digit Diagnostic Code 1.999. En consulta de 10/09/2020, Google Académico refiere 398 citas.
3.- The Institute of Medicine (IOM) -Stratton1.9963 Tiene su origen en una orden del Congreso de los EEUU al IOM para el estudio de herramientas para el diagnóstico, la prevalencia y la eficacia de los programas de prevención y tratamiento. En consulta de 10/09/2020, Google Académico refiere 1.354 citas.
4.- Diagnostic Guide for Fetal Alcohol Syndrome and Related Conditions: The 4-Digit Diagnostic Code 1.9994-5. La primera se desarrolló en 1.997. No la incluimos por no haber podido tener acceso. La que tratamos supone la segunda edición y se desarrolló por el equipo del Diagnostic and Prevention Network (FAS DPN) de la University de Washington a partir de los datos de 1.014 pacientes. Su objetivo es el de superar las limitaciones, según los propios autores, de las guías que hemos descrito anteriormente. Tiene el soporte y contribución de Centers for Disease Control and Prevention; Center on Human Development and Disability, University of Washington, Seattle WA; Division of Alcohol and Substance Abuse, Washington State Department of Social and Health Services; March of Dimes Birth Defects Foundation y John B. Chavez FAS Fund. En consulta de 02/09/2020, Google Académico, refiere 166 citas para el primer documento y 556 para el segundo.
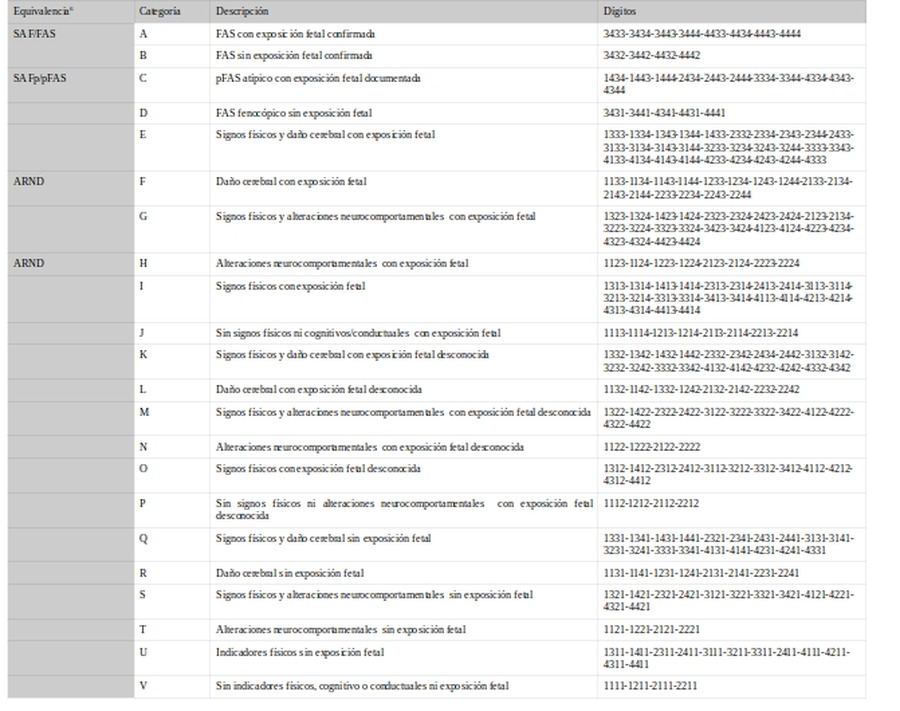
Se basa en 4 componentes diagnósticos que son valorados por escala.
A partir de los 4 dígitos establecen 22 categorías diagnóstica:
5.- Emory-20 2.0007 No hemos podido disponer de los trabajos fuente, sí que tiene su origen en Emory-Fetal Alcohol Center Clinical Criteria, Atlanta, Georgia. El trabajo del que hemos extraído la información obtiene en consulta de 02/09/2020 a Google Académico, 69 citas.
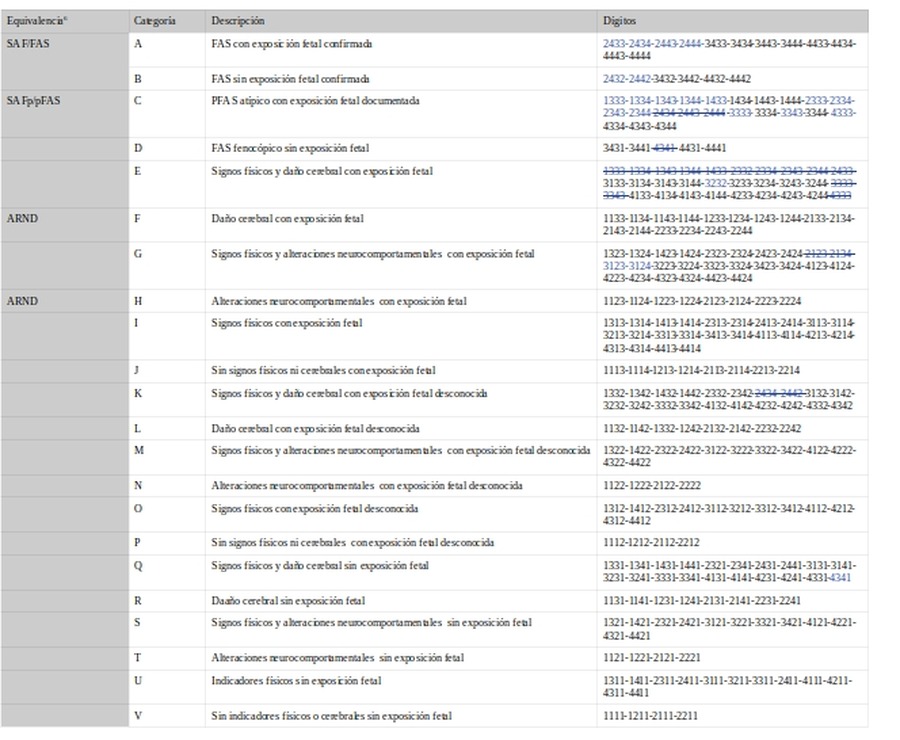
6.-Diagnostic Guide for Fetal Alcohol Syndrome and Related Conditions: The 4-Digit Diagnostic Code 20048. Supone una revisión y actualización de la de 1.999. Los cambios introducidos son, según los autores:
a.- Guía labio-filtrum: se añade la correspondiente a los afro-americanos. Para los caucasianos se ajusta la circula ridad para el rango 4.
b.- Criterio del peso/altura: en esta actualización se considera signo el rango 2 (<=10 percentiles) mientras que en el anterior se requería un peso/altura inferior al rango 3 (<=3 percentiles). Igualmente se ha actualizado el gráfico de peso/altura al de 2.000 del Centers for Disease Control and Prevention.
c.- Reclasificación de 19 de los códigos, mayoritariamente por los cambios de criterio en relación a peso/altura. Tales cambios los hemos marcado en azul para facilitar la lectura.
En consulta de 10/09/2020, Google Académico, refiere 8 citas.
A partir de los 4 dígitos establecen 22 categorías diagnóstica (en azul los cambios respecto a versión anterior):
7- Centers for Disease Control and Prevention (CDC)-2.0049
Desarrollado por un grupo de expertos dentro del Centers for Disease Control and Prevention (Department of Health and Human Services) en coordinación con National Task Force on Fetal Alcohol Syndrome and Fetal Alcohol Effect American, Academy of Pediatrics, American College of Obstetricians and Gynecologists, March of Dimes y National Organization on Fetal Alcohol Syndrome. Fue la respuesta al mandato que el Congreso de los EEUUU les hizo en 2.002. En consulta de 02/09/2020, Google Académico, refiere 255 citas.
8.- Canadian diagnostic guideline 2.00510 La versión inicial de 2.005 fue desarrollada por expertos en el diagnósticos de Canada y EEUU. Suponía una síntesis entre las 4 categorías diagnósticas IOM y la clasificación The 4-Digit Diagnostic Code. En consulta de 10/09/2020, Google Académico refiere 1.008citas.
9.- The Institute of Medicine (IOM) –Hoyme 200511 Se elaboró a partir de una cohorte de niños con EPA/PAE de 6 comunidades de nativos americanos y una comunidad de Sudáfrica. Para los autores supone una concreción para la práctica clínica de la de 1.996 Como aproximación bibliométrica informar que, en consulta de 02/09/2020, Google Académico refiere 974 citas.
10.- German guideline version 201312. Fue desarrollada por un grupo de expertos con revisión bibliográfica y valoración del grado de evidencia siguiendo el Oxford Classification System 2009. El Ministerio Federal de Salud de Alemania (GFMOH) realizó la financiación. En consulta de 02/09/2020, Google Académico informa de 46 citas.
11.- Canadian diagnostic guideline 201513 Supone una evolución de la del 2.005 de forma que se abandonan las categorías IOM y se diferencia entre FASD con y sin rasgos faciales (SFF),, se incluye la categoría no diagnóstica “Riesgo de FASD” y se elimina el criterio de alteraciones del crecimiento. En consulta de 10/09/2020, Google Académico, refiere 279 citas.
12.- Australian Guide to the diagnosis of Fetal Alcohol Spectrum Disorder (FASD)-201614-15 Ha sido desarrollada por un grupo de expertos y financiado por el Commonwealth Department of Health (DoH). Su primera versión es del 2.012. Consideran que sus criterios son similares a la Canadiense de 2.015 e informan que utilizan la guía de Astley para la evaluación de dismorfologías faciales del 2.004. En consulta de 02/09/2020, Google Académico informa de 19 citas para el artículo.
13.- The Institute of Medicine (IOM) –Hoyme 201616. Supone una actualización de la del 2005. Se basó en una revisión de la bibliografía, en la experiencia de los autores con más de 10.000 niños valorados en entorno clínico y estudios epidemiológicos. Se trata de un trabajo conjunto del “National Institute on Alcohol Abuse and Alcoholism– funded studies, the Collaborative Initiative on Fetal Alcohol Spectrum Disorders, and the Collaboration on FASD Prevalence”. En consulta de 02/09/2020, Google Académico, refiere 284 citas.
14.- Por su parte Mattson et al. -201917 añade a los criterios The Institute of Medicine (IOM) -Hoyme 2016 los Trastornos Neurocomportamentales Relacionados con la Exposición Prenatal al Alcohol (ND-PAE). Se trata de una recomendación que fue publicada en el último DSM5, pero que ha sido discutida y mayoritariamente poco utilizada (O. García Algar, comunicación personal 06/08/2020). En consulta de 02/09/2020, Google Académico, refiere 33 citas.
Finalmente y para acabar de contextualizar estos criterios exponemos la frecuencia de su uso en estudios epidemiológicos obtenidos del metanálisis de Lange et al18 para el periodo 1.973-2.015 sin restricciones geográficas o de idioma:
En una entrada anterior hablábamos de la mayor vulnerabilidad biológica del feto masculino y su implicación en la mayor prevalencia en la discapacidad intelectual (DI). También hemos tratado sobre la exposición prenatal al alcohol (EPA//PAE) en relación a la prevalencia, muerte fetal (FD) e interrupción voluntaria del embarazo tras diagnóstico de malformación fetal (TOPFA). Nuestra hipótesis inicial es la de una mayor prevalencia masculina.
Las primeras observaciones de que disponemos sobre el Índice de Masculinidad (IM) en el Síndrome Alcoholico Fetal (SAF/FAS) son del 1.9761. En ellas, con una muestra de 64 SAF/FAS, se observa un IM de 0,52. Dado que las edades estaban entre 1 y 7 años, los mismos autores, manifiestan que podría deberse a mayor muerte fetal (FD) o en primeras etapas extrauterinas.
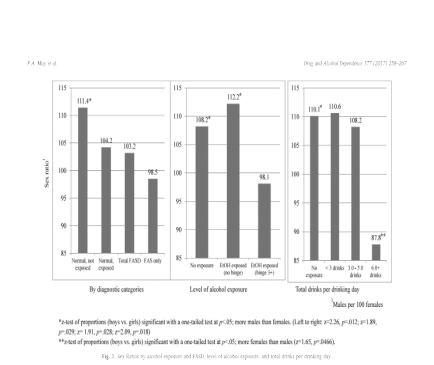
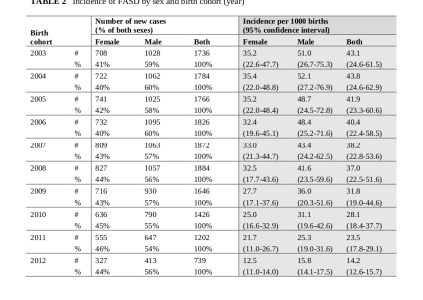
En el trabajo de May et al2 se observaron los siguientes resultados:
Los autores entienden, como los primeros, que el menor indice de masculinidad en SAF/FAS (0,98) y Trastornos del espectro del alcoholismo fetal (TEAF/FASD) (1,03) respecto al de la población de referencia (1,11) se debería a una mayor mortalidad pre y perinatal en varones. En el tercer gráfico podemos observar que parece que tal susceptibilidad es proporcional a la cantidad de ingesta. Sin embargo, como señalábamos en una entrada anterior, los datos EUROCAT33 infieren buena viabilidad fetal y baja mortalidad perinatal. Por otro lado tenemos el trabajo de García et al3a en el que analizaron el meconio en 353 casos diferenciando entre alto consumo frente a ocasional o abstinencia (>=2nmol/gr / <2nmol/gr ). En este se observó un IM para el grupo de madres abstinentes u ocasionales de 0,98 (n=194) frente a 1,52 (n=159) sin significación estadística.
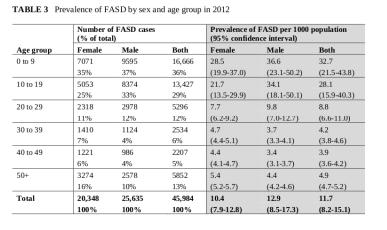
Interesantes son los datos de Thanh et al4 del que extraemos las siguientes tablas:
Y son interesantes por varios motivos. En primer lugar parecen constatar la mayor mortalidad masculina pero no perinatal. En segundo lugar, no apoya la tesis de mayor prevalencia entre mujeres. En último lugar, muestra una significativa fluctuación en pocos años en un mismo lugar.
De la lectura de diversos trabajos sobre el tema hemos obtenido la impresión de que si bien FAS y FASD pueden entenderse como un continuum cuantitativo son cualitativamente realidades distintas. En otra entrada intentaremos concretar más este punto. Por el momento lo que haremos es centrarnos en FAS.
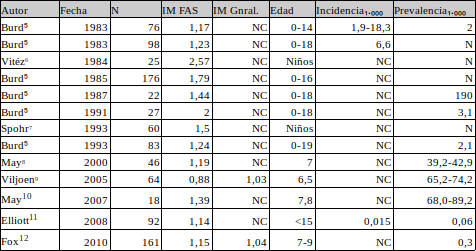
Para ello hemos revisado los artículos que se obtienen en Google Académico con “fetal alcohol syndrome prevalence sex age” y agrupado en la tabla siguiente. Somos conscientes de las limitaciones que esta metodología y de que los resultados que podamos obtener se han de considerar una primera aproximación.
Puede observarse que los IM encontrados oscilan entre 0,88 y 2,57 y una media ponderada de 1,35. Ello nos lleva a mantener la hipótesis inicial de mayor incidencia masculina y que la mayor vulnerabilidad biológica de este grupo también se manifiesta en teratogénia por alcohol.
1.- Qazi, Q. H., & Masakawa, A. (1976). Altered sex ratio in fetal alcohol syndrome. Lancet (London, England), 2(7975), 42. 2.- May, P. A., Tabachnick, B., Hasken, J. M., Marais, A. S., de Vries, M. M., Barnard, R., … & Buckley, D. (2017). Who is most affected by prenatal alcohol exposure: Boys or girls?. Drug and alcohol dependence, 177, 258-267. 3.- https://eu-rd-platform.jrc.ec.europa.eu/eurocat/eurocat-data/prevalence/export/, accessed on 7/6/2020. 3.a.- Garcia-Algar, O., Kulaga, V., Gareri, J., Koren, G., Vall, O., Zuccaro, P., … & Pichini, S. (2008, April). Alarming prevalence of fetal alcohol exposure in a Mediterranean city. In Therapeutic drug monitoring (Vol. 30, No. 2, pp. 249-254). LWW. 4.- Thanh, N. X., Jonsson, E., Salmon, A., & Sebastianski, M. (2014). Incidence and prevalence of fetal alcohol spectrum disorder by sex and age group in Alberta, Canada. Journal of Population Therapeutics and Clinical Pharmacology, 21(3). 5.- h.- Burd, L., & Moffatt, M. E. (1994). Epidemiology of fetal alcohol syndrome in American Indians, Alaskan Natives, and Canadian Aboriginal peoples: a review of the literature. Public Health Reports, 109(5), 688. 6.- Vitéz, M., Korányi, G., GŌNCZY, E., Rudas, T., & Czeizel, A. (1984). A semiquantitative score system for epidemiologic studies of fetal alcohol syndrome. American journal of epidemiology, 119(3), 301-308. 7.- Spohr, H. L., Willms, J., & Steinhausen, H. C. (1993). Prenatal alcohol exposure and long-term developmental consequences. The Lancet, 341(8850), 907-910. 8.- May, P. A., Brooke, L., Gossage, J. P., Croxford, J., Adnams, C., Jones, K. L., … & Viljoen, D. (2000). Epidemiology of fetal alcohol syndrome in a South African community in the Western Cape Province. American journal of public health, 90(12), 1905. 9. Viljoen, D. L., Gossage, J. P., Brooke, L., Adnams, C. M., Jones, K. L., Robinson, L. K., … & Asante, K. O. (2005). Fetal alcohol syndrome epidemiology in a South African community: a second study of a very high prevalence area. Journal of studies on alcohol, 66(5), 593-604. 10.- May, P. A., Gossage, J. P., Marais, A. S., Adnams, C. M., Hoyme, H. E., Jones, K. L., … & Hendricks, L. (2007). The epidemiology of fetal alcohol syndrome and partial FAS in a South African community. Drug and alcohol dependence, 88(2-3), 259-271. 11.- Elliott, E. J., Payne, J., Morris, A., Haan, E., & Bower, C. (2008). Fetal alcohol syndrome: a prospective national surveillance study. Archives of Disease in Childhood, 93(9), 732-737. 12.- Fox, D. J., Pettygrove, S., Cunniff, C., O’Leary, L. A., Gilboa, S. M., Bertrand, J., … & Frías, J. L. (2015). Fetal alcohol syndrome among children aged 7–9 years—Arizona, Colorado, and New York, 2010. MMWR. Morbidity and mortality weekly report, 64(3), 54.
Bajo la denominación de Trastornos del Espectro Alcohólico Fetal (TEAF/FASD) se agrupan un conjunto de signos y síntomas que se presentan como consecuencia de la exposición prenatal al alcohol (EPA/PAE). Incluye:
a.- Síndrome alcoholico fetal (SAF/FAS): alteraciones del crecimiento físico, dismorfología (especialmente facial), afectación cognitiva y problemas de conducta.
b.- Síndrome alcoholico fetal parcial (SAFP/PFAS): destaca una menor alteración del crecimiento.
c.- Efectos del alcoholismo fetal (EAF/FAE) que se desglosa en:
c.1.- Trastornos Congénitos Relacionados con el Alcohol (ARBD): destacan las alteraciones estructurales y dismórficas con alteraciones cognitivas y conductuales menores.
b.2.- Trastornos del Neurodesarrollo Relacionados con del alcohol (ARND): destacan las alteraciones cognitivas y conductuales con menor dismorfología.
En el trabajo de Evrard1 podemos encontrar la descripción, criterios diagnósticos y diferenciales. Por nuestro ámbito de interés destacamos, según el mismo autor, que se trata de la principal causa teratógena de discapacidad intelectual (DI). Sin embargo, el análisis de los datos epidemiológicos se ve complicado por el hecho de que si bien, con gran certeza, se van a presentar alteraciones neuropsicopatológicas no se va a poder deducir que se cumplen los criterios diagnósticos para la discapacidad intelectual. Otra dificultad vendrá dada por que la percepción de consumo de alcohol en general (y durante el embarazo) variará de acuerdo a criterios culturales, geográficos y sociales. El mismo concepto de consumo es difícil de definir y medir.
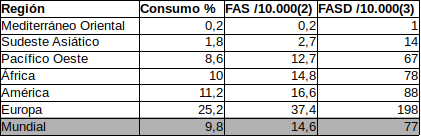
Popova et al2 realizaron un metanálisis en que se observó una tasa de consumo de alcohol (>= 2 consumiciones/día o 5-6 consumiciones/episodio) en el embarazo del 9,8% mundial con el nivel más bajo del 0,2% en el Mediterráneo Oriental y del 25,02% en Europa. Por su parte, Lange et al3 y para el mismo año (2.012) realizaron un metaánalisis para TEAF/FASD. En la tabla siguiente se muestran los datos y puede observase la correlación con la prevalencia de SAF/FAS y TEAF/FASD.
Fuente: elaboración propia a partir de 2 y 3.
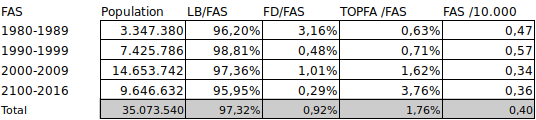
Por nuestra parte, a partir delos datos de la European network of population-based registries for the epidemiological surveillance of congenital anomalies (EUROCAT)4hemos obtenido la serie histórica 1980-2016 referente a embarazos y nacimientos que hemos agrupado, para simplificar, en la siguiente tabla.
FAS: Síndrome alcoholico fetal. LB: Nacidos vivos. FD: muerte fetal o nacidos muertos tras 20 semanas de gestación.
TOPFA: interrupción voluntaria del embarazo tras diagnóstico de malformación fetal.
La comparación de la incidencia al nacimiento (0,40/10.000) con la prevalencia ya señalada (37,4/10.000) descarta una mortalidad prematura significativa. Así se ha observado una esperanza de vida de 34 años y las siguientes tasas de mortalidad:
Edad
% SAF/FAS5
% S Down
0
7,76
20,06
1-4
0,89
5,336
5-9
0,37
0,196
10-14
0,20
0,217
15-19
0,95
0,197
20-24
3,34
0,317
25-29
2,47
0,47
30-34
3,60
0,597
35-39
3,61
0,637
40-44
10,26
1,077
45-49
8,96
2,627
50-54
6,25
3,757
55-59
18,18
5,647
60+
21,43
12,257
Fuente: elaboración propia a partir de las fuentes señaladas. Debe tenerse presente las diferencias temporales y geográficas. El propósito de los datos sobre S Down son los de permitir un punto de referencia.
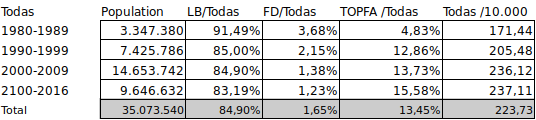
Hemos querido observar el comportamiento pre y perinatal de este síndrome respeto de otras. Para ellos hemos tomado como referencia el total de las 92 anomalías congénitas de la misma fuente 4:
De la comparación de ambas tablas se observa, en primer lugar, que el SAF/FAS presenta una buena viabilidad fetal. Así el total de muertes fetales o nacidos muertos representa el 0,92% frente al 1,65% para el total de anomalías congénitas. Diferencia que pensamos sería mayor dado que una parte de las TOPFAs corresponderían a fetos que hubiesen podido acabar como FD en mayor número para el total que para FAS.
En segundo lugar, se observa una diferencia importante de TOPFA (1,76% para SAF/FAS frente a 13,45% para el total). Tal diferencia podría venir por la ausencia de diagnóstico prenatal específico junto a la baja presencia de defectos estructurales graves en el FAS.
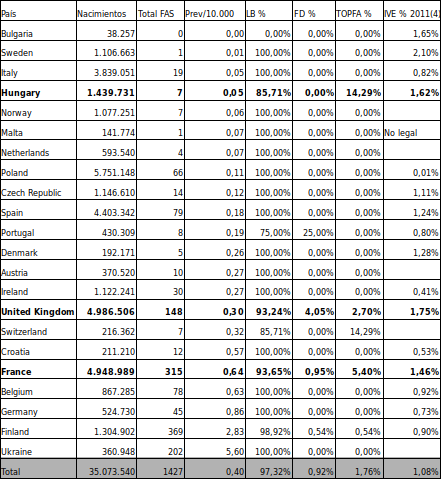
La tabla siguiente, elaborada a partir de la fuente de datos ya mencionada, nos permite comparar la frecuencia de TOPFA por SAF/FAS con la tasas de interrupción por cualquiera de las causas contempladas en la legislación por cada 100 mujeres entre 15 y 44 años (año 2011)8.
Fuente: elaboración propia a partir de 4 y 8
Dada la variabilidad de supuestos y plazos entre países8 pensamos que son estos los que más peso tendrían a la hora de explicar las diferencias de TOPFA. Así haría más referencia a la decisión de interrumpir el embarazo por causas personales que por malformaciones SAF/FAS.
Debemos señalar que no parecen haber diferencias en prevalencia por raza, etnia o nivel socioeconómico ante igual consumo9..
Para finalizar la entrada señalar que los TEAF/FASD constituyen un conjunto diagnóstico complejo y sobre el que no hay consenso. Ello será tratado en otras entradas.
1.- Evrard, S. G. (2010). Criterios diagnósticos del síndrome alcohólico fetal y los trastornos del espectro del alcoholismo fetal. Arch Argent Pediatr, 108(1), 61-67.
2.- Popova, S., Lange, S., Probst, C., Gmel, G., & Rehm, J. (2017). Estimation of national, regional, and global prevalence of alcohol use during pregnancy and fetal alcohol syndrome: a systematic review and meta-analysis. The Lancet Global Health, 5(3), e290-e299.
3.- Lange, S., Probst, C., Gmel, G., Rehm, J., Burd, L., & Popova, S. (2017). Global prevalence of fetal alcohol spectrum disorder among children and youth: a systematic review and meta-analysis. JAMA pediatrics, 171(10), 948-956.
4.- https://eu-rd-platform.jrc.ec.europa.eu/eurocat/eurocat-data/prevalence/export/, accessed on 7/6/2020.
5.- Thanh, N. X., & Jonsson, E. (2016). Life expectancy of people with fetal alcohol syndrome. Journal of Population Therapeutics and Clinical Pharmacology, 23(1).
6.- Day, S. M., Strauss, D. J., Shavelle, R. M., & Reynolds, R. J. (2005). Mortality and causes of death in persons with Down syndrome in California. Developmental Medicine & Child Neurology, 47(3), 171-176.
7.- Fryers, T., & Mackay, R. I. (1979). Down syndrome: prevalence at birth, mortality and survival. A 17-year study. Early Human Development, 3(1), 29-41.
8.- Domínguez, J. N. (2014). Análisis de la legislación europea y española sobre salud sexual y reproductiva. Revista Fundación Alternativas.
9.- May, P. A., Hasken, J. M., Stegall, J. M., Mastro, H. A., Kalberg, W. O., Buckley, D., … & Tabachnick, B. G. (2020). Fetal alcohol spectrum disorders in a southeastern county of the United States: child characteristics and maternal risk traits. Alcoholism: Clinical and Experimental Research, 44(4), 939-959.








{kind=link}